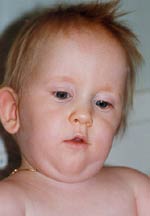
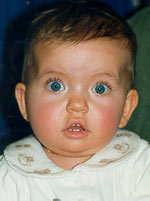
Klinik und Genetik des Prader-Willi Syndroms
Univ. Doz. Dr. Olaf Rittinger / Salzburg
aus: Klinische Pädiatrie 2001; 213: 91-98
Zeitschrift für Klinik und Praxis
Sonderdruck
| Das Prader-Willi Syndrom (PWS) zählt zu den seltenen neurogenetischen Funktionsstörungen und stellt andererseits die häufigste Form des syndromatischen Übergewichts dar. Klinische Hauptmerkmale sind eine im Neugeborenenalter ausgeprägte Hypotonie mit anfänglich erheblicher Trinkschwäche und dadurch bedingter Gedeihstörung, ab dem 2. Lebensjahr Entwicklung einer Hyperphagie mit stammbetonter, unbehandelt bisweilen morbider Adipositas. Neben einer verzögert ablaufenden motorischen Entwicklung ist auch der Spracherwerb verlangsamt und bleibt oft eingeschränkt, die intellektuelle Entwicklung ist durch eine Lernbehinderung mit Beeinträchtigung besonders des Kurzzeitgedächtnisses und abstrakter Denkvorgänge gekennzeichnet.
Der genetische Hintergrund aller bisher bekannter Genotypen ist ein Funktionverlust eines väterlich vererbten Genclusters am Chromosom 15 (q11.2). Die elternspezifische Prägung (genomic imprinting) stellt ein epigenetisches Phänomen dar, wo-durch identische Basensequenzen homologer elterlicher Gene ein- und ausgeschaltet werden können. Das PWS wie auch das molekulargenetisch eng benachbarte, klinisch allerdings völlig andersartige Angelman-Syndrom (AS) zählen zu den ersten Krankheitsbildern, bei denen ein Funktionsausfall eines Imprint-Gens als Krankheitsursache nachgewiesen wurde. Bei unterschiedlichen Genotypen ist der Phänotyp vergleichsweise sehr ähnlich und zeigt beim PWS nur diskrete statistische Differenzen, z.B. bezüglich der Pigmentierung. Eine frühe klinische Diagnose ist in Kenntnis des neuromuskulären Defizits anzustreben, weil eine frühzeitig einsetzende gut strukturierte kalorienreduzierte Ernährung dem sonst ungehemmten Gewichtszuwachs mit nachfolgenden Komplikationen entgegenwirken kann. Die Diagnose über die Morphologie allein ist in der Neugeborenenperiode schwierig und wird oft erst durch die Kombination mit der erheblichen Hypotonie in Betracht gezogen. Die Verfügbarkeit verläßlicher, mittlerweile bevorzugt nicht-radioaktiver molekulargenetischer Nachweismethoden läßt die Diagnose zweifelsfrei feststellen und ein tragfähiges Konzept für eine sinnvolle symptomatische Therapie erarbeiten. Zu diesen Maßnahmen zählt auch der Einsatz von rekombinantem Wachstumshormon, das nach den Ergebnissen zahlreicher klinischer Studien bei gegebenem funktionellen Wachstumshormonmangel einen zweifelsfreien auxologischen Nutzen und auch eine günstige Veränderung der Fett bzw. Muskelmasse und eine Verbesserung der motorischen Aktivität und der respiratorischen Funktion bewirkt. Zusammenfassung: Eine frühzeitige Diagnose des PWS mittels DNA-Untersuchung verhilft zu einer klaren Auskunft hinsichtlich der Genetischen Beratung, erspart dem Kind unnötige zusätzliche Untersuchungen wie CT und Muskelbiopsie und bereitet den Weg zu einer gezielten symptomatischen Therapie, zu der im Hinblick auf den funktionellen HGH-Mangel bei PWS auch eine Wachstumshormontherapie empfehlenswert erscheint. Wegen der im Verlauf der Entwicklung vielfachen möglichen Komplikationen erscheint ein multidisziplinäres Betreuungskonzept an einem Zentrum empfehlenswert. |
Phänotyp unterschiedlicher Genotypen:
|